HyperChem �n��\���
�@
NEW HYPERCHEM 8.0! MOLECULAR MODELING SOFTWARE FOR
WINDOWS. CREATE AND DO CALCULATIONS ON MOLECULES. CALL FOR NETWORK OR UPGRADE
PRICING
HyperChem is a sophisticated molecular modeling environment that is known for
its quality, flexibility, and ease of use. Uniting 3D visualization and
animation with quantum chemical calculations, molecular mechanics, and dynamics,
HyperChem puts more molecular modeling tools at your fingertips than any other
Windows program.
Our newest version, HyperChem Release 8.0, is a full 32-bit application,
developed for the Windows 95, 98, NT, ME, 2000, XP, and Vista operating systems.
HyperChem Release 8.0 incorporates even more powerful computational chemistry
tools than ever before, as well as supporting multiple third-party applications.
Its drawing and rendering capabilities and ease of use are standards for the
industry.

What is HyperChem?
HyperChem is a sophisticated molecular modeling environment that is known for
its quality, flexibility, and ease of use. Uniting 3D visualization and
animation with quantum chemical calculations, molecular mechanics and dynamics,
HyperChem puts more molecular modeling tools at your fingertips than any other
Windows program. It includes all the components of structure, thermodynamics,
spectra, and kinetics.

What is New in HyperChem 8?
HyperChem 8.0 is the latest release of HyperChem. It introduces extensive new
features that add to its industry-leading position. This is particuarly true
with regard to its "open architecture" and HyperChem's ability to interact with
other programs such as Excel and Word or third-party packages in computational
chemistry.
Third-Party Interfaces In spite of the fact that HyperChem has extensive
and elegant number crunching capabilities, it can't be all things to all
computational chemists. With Release 8, an effort to enable HyperChem as a
universal component in computational chemistry begins. Release 8 includes an
interface to six third-party packages that are in common use in computational
chemistry. None of these packages includes a native graphical user interface.
Each benefits from the capabilities of HyperChem and each set of users now has
capabilities never imagined before. Now PC Gamess, WinGamess, PQS, Q-Chem,
Gaussian, and Mopac2007 can all be transparently executed by HyperChem. Now each
of these can act as a computational engine for HyperChem. They add to the large
number of native computational engines already in HyperChem, such as HyperGauss,
HyperDFT, etc. The world just got easier.
Other Additions
- Double precision everywhere
- New Geometry measurements (POINT to PLANE, etc)
- Additions to model builder (substituents)
- New semi-empirical RM1 method
- Render POINT, LINE, and PLANE
- Calculation of Entropies, Heat Capacities, FreeEnergies
- Calculation of zero-point energies
- Additions to toolbar (C, N, O, etc), and Model Builder
- Undo and Redo of Model Builder
- A recent file list has been added for ease of use
- CI separated from SCF dialog box
- Batch calculations which spawn back ends and return
- Vista compatibility
- Line width envelopes for IR and UV spectra
- Vibrations for molecular mechanics
- Named FIXEDATOMS and MECHANICALATOMS
- Expanded use of MP2
- Calculation of Equilibrium Constants w/ Temperature
- Electric fields for molecular mechanics
- Calculation of Kinetic Rate Constants w/ Temperature
- Options for units (kCal, kJ, or a.u.)
- Calc. of particle-in-box energy and wave function

HyperChem Release 8.0 Feature Summary
Structure Input and Manipulation
Building molecules with HyperChem is simple: just choose an element from the
periodic table, and click and drag with the mouse to sketch a structure as you
would on paper, prior to applying the model builder to convert your 2D sketch to
a 3D structure. Alternatively, select Hydrogen atoms and substitute them with a
variety of substituents including your own custom substituent. If you make a
mistake, just push the Undo button. Mouse control of rotation around bonds,
stereochemistry, and "rubber banding" of bonds makes changing structures easy.
Extensive selection, highlighting, and display capabilities make it easy to
focus on areas of interest in complex molecules.
- Select, rotate, translate, and resize structures with convenient
mouse-controlled tools. Modify settings to control operation of the tools.
- Convert rough sketches into full 3D structures with HyperChem's advanced model
builder.
- Apply builder constraints easily: specify bond lengths, bond angles, torsion
angles, or the bonding geometry about a selected atom.
- Specify atom type, formal charge, atomic charge, atomic mass, and custom atom
labels.
- Build clusters and complex molecular assemblies; move individual atoms and
molecules as easily as you move groups; automate molecular design with scripts;
create complicated molecular systems.
- Just click to build peptides and nucleic acids from amino acid and nucleotide
residue libraries; add counter ions or create zwitterions; inherit charges from
templates for Amber, Charmm, and OPLS.
- Mutate residues and build large molecules incrementally (make changes at any
point).
- Cut and paste protein segments for homology modeling; model proteins with over
100,000 atoms.
- Read protein sequences and create specific secondary structure as input to
modeling calculations.
- Add a periodic box of pre-equilibrated water molecules for aqueous solvation
studies. Periodic boundary conditions can be used with other solvent systems, or
without solvents.
- Import structures from standard file formats: Protein Data Bank PDB, ChemDraw
CHM, MOPAC Z-matrix, MDL MOL and ISIS Sketch, Tripos MOL2 files, and Cambridge
Crystallographic files.
- Build molecular crystals from individual molecules; set crystal type and unit
cell parameters.
- Create atomic crystals with a variety of sample unit cells, with or without
periodic boundary conditions.
- Use a library of aldoses, ketoses, and derivatives to automate the building of
polysaccharides from components; create your own sugar components as desired.
- Invoke the polymer builder to build straight or branched polymers from
arbitrarily drawn monomers; specify all the structural parameters for monomer
linkages or optionally randomize linkages.
- Input molecules directly from a database of your own creation or choose from
over 10,000 molecules in HyperChem's database.
- Perform a Root-Mean-Square (RMS) fit of one molecule to another.
- Assign formal charges to atoms for display and for deciding on how many
hydrogens the model builder will add (i.e. specify the valence).
- Create a custom component with one selected Hydrogen and store it away. Later
use it as a substituent, R, anywhere you like or use common pre-defined
substituents instead. Every option for creating your molecule is available.
- Made a mistake altering a structure of deleting some atoms? Just press Undo.
Changed your mind? Press Redo.
Molecular Display
- Display structures using sticks, ball and stick, ball and cylinders, fused CPK
spheres, or tubes; choose any rendering for any atom; switch easily between
rendering styles; quickly center any selected portion of a molecule.
- Add a dot surface to any atom; choose your own stick width or the radius of
balls, cylinders and tubes.
- Specify your own colors for simple, repetitive use; select from any of 16M
colors.
- Show orbitals and electron densities as 2D contours or 3D isosurfaces.
- Display molecular electrostatic potential as 3D isosurface or as mapped onto
electron density surface.
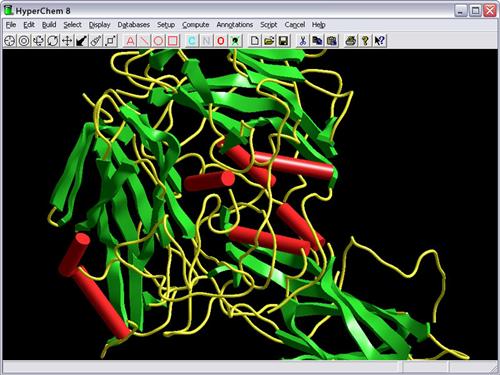
- Display protein backbones with optional display of sidechains; display
high-quality secondary structure renderings such as a cylinder for a helix, a
thick ribbon for a beta sheet or tubing for a random coil.
- Use selection to mix and match what is shown and how; distinguish atoms versus
secondary structures.
- Highlight potential hydrogen bond interactions and render accordingly.
- Display dipole moment vectors or vectors representing the force on any or all
atoms.
- Show aromatic rings via either of two user-selected methods.
- Produce ray-traced graphics from HyperChem structures; assign a variety of
lights with different position and color.
- Display atom labels such as symbol, name, number, type, charge, spin, mass,
basis set, chirality, and gradient.
- Display bond labels such as length and bond order or residue labels such as
type and number.
- Display custom user-defined atom labels as annotations to a structure.
- Display inertial axes and moments of inertia for any selected portion of a
molecular system.
- Display POINT, LINE, and PLANE in a graphical way as structural features of
any molecule or selection.
- Display periodic boundary conditions
- Hide any selected portion of system.
- Display overlap of two molecular systems as per their RMS fit.
- Use a variety of user selected colors for window backdrops, atoms, selections,
orbitals, and isosurfaces; color portions of a system according to your own
needs and tastes.
- Display/Edit molecular mechanics or semi-empirical parameters for selected
atoms, bonds, and angles.
- Copy displayed images to file or clipboard for incorporation into another
program such as Microsoft PowerPoint or for display on a web page.
- Capture structure, orbitals, or spectra and immediately create an active web
page allowing rotation of molecule, inspection of orbitals, or animation of
normal mode; freely distribute web control to allow others to view your active
content.
- Apply and display names for individual molecules of a system.
- Immediately display potential energy surfaces for one or two independent
structural variables; save as potential energy plot for presentations.
- Display spectra including frequency, intensity and line-width envelopes. Click
on any line for further information.
- Display vectors attached to atoms indicating the relative force on the atom or
its direction of motion in a normal mode.
- Display animations of normal modes from vibrational analysis (IR Spectra).
Selections
- Apply most operations either to the current selection or alternatively to the
whole system.
- Select and name sets of atoms for custom display or monitoring of properties.
- Select a set of atoms and apply pre-defined properties POINT, LINE, PLANE,
REACTANT, PRODUCT, PLOT1, PLOT2, GHOST ATOMS, NMR ATOMS, FIXED ATOMS, MECHANICAL
ATOMS, and QUANTUMATOMS to the atoms.
- Select with unit of selection being atoms, residues, or whole molecules.
- Select atoms based on their individual properties - such as all atoms within a
specified range of energy gradient.
- Select atoms associated with a secondary structure such as beta-turn atoms,
helix atoms, etc.
- Logically complement a selection.
Annotations
- Draw text, lines, ellipses and rectangles (filled or unfilled) on annotation
layer to document your modeling results; apply z-ordering to your annotations
for proper visual effect. In essence, a small general drawing program is include
in HyperChem.
- Create symbol annotations as text in a particular font and size.
- Hide annotation layer or place it in front of or behind modeling layer.
- Save annotations in same file as molecular system to save/restore a
general-purpose drawing or a specific set of annotations.
- Select and zoom/translate annotations as you would atoms of a molecule.
- Draw the bottom of a one-dimensional box and see the "particle-in-a-box"
energies and wave functions.
Database
HyperChem 8.0 includes a separate database package that interacts intimately
with HyperChem to provide molecules for input to modeling calculations and to
store results of those calculations.
- Create a new database of 2D molecular drawings with associated 3D structures
and molecular data.
- Open a default existing database of over 10,000 common molecules.
- Create a new database record from HyperChem with the push of a button.
- Create 2D drawings for publication and presentation purposes.
- Query a database for 2D structure and values of the database fields.
- Run a script over a database of molecules to automate HyperChem calculations.
- Create a general-purpose 3D query but associating query with (true, false)
result of any HyperChem script.
- Create a database of molecules to carry around on your phone or Pocket PC.
- Use standard Microsoft Access *.mdb database files.
- Convert 2D structures to 3D structures or vice versa.

Computational Chemistry
Use HyperChem to explore quantum or classical model potential energy surfaces
with single point, geometry optimization, or transition state search
calculations. Include the effects of thermal motion with molecular dynamics,
Langevin dynamics or Metropolis Monte Carlo simulations. User-defined structural
restraints may be added. Use translational, rotational, and vibrational
partition functions to calculate entropies, heat capacities, and free energies.
Types of Calculations
- Single point calculations determine the molecular energy and properties for a
given fixed geometry; SCF, configuration interaction, or MP2.
- Geometry optimization calculations employ energy minimization algorithms to
locate stable structures. Six minimization algorithms are provided. Even
calculate geometries with MP2 or for excited states.
- Vibrational frequency calculations find the normal vibrational modes of an
optimized structure. The vibrational spectrum can be displayed (with IR
intensities) and the vibrational motions associated with specific transitions
can be animated.
- Electronic spectra with visual display of frequencies and intensities.
- Transition state searching locates the metastable structures corresponding to
transition states using either Eigenvector Following or Synchronous Transit
methods. Molecular properties are then calculated.
- Molecular dynamics (MD) simulations compute classical trajectories for
molecular systems. Quantum forces can be used to model reactive collisions.
Heating, equilibration, and cooling periods can be employed for simulated
annealing and for studies of other temperature-dependent processes. Both
constant energy and constant temperature simulations are available.
- Langevin dynamics simulations add frictional and stochastic forces to
conventional molecular dynamics to model solvent collisional effects without
inclusion of explicit solvent molecules.
- Metropolis Monte Carlo simulations sample configurations from a statistical
ensemble at a given temperature and are useful for exploring the possible
configurations of a system as well as for computing temperature-dependent
equilibrium averages.
- Calculate the energy and entropy at a temperature T using an analysis of the
translation, rotation, and vibration of a molecule. Subsequently obtain the free
energy as A=E-TS.
- Calculate the equilibrium constant for any reaction and plot as a function of
the temperature.
- Calculate the rate constant for unimolecular and bimolecular reactions from
the HyperChem (or other) structures obtained for the transition state and the
reactant species. Plot the rate constant as a function of temperature.
- Calculate heat capacities for molecules at temperature T from their
translation, rotation, and vibration.
- Apply a superimposed electric field to any molecular system. See what changes
this makes to any molecular modeling result such as structure, energetics, and
reactivity.
- Apply a superimposed magnetic field to a molecular system for a subset of the
semi-empirical methods.
- Specify that your results be described by any of the three system of units -
kJ, kCal, or atomic units(Hartrees.).
- Calculations generally apply to any applicable method, e.g. - ab initio
- MD or vibrational spectra with DFT, etc.
- QSAR properties (Gasteiger charges, surface area (2 methods), volume,
hydration energy, log P, refractivity, sum of bond polarizabilities, mass).
- Conformational search (Monte Carlo generation of conformers, subsequent
optimization, and simple collection of table of low energy conformers); easy
inspection and manipulation of each conformer.
- NMR shielding and coupling constants with optional subsequent computation and
display of 1-D spectra; special TNDO technology gives rapid computation of NMR
parameters with improved accuracy over other semi-empirical methods.
Ab Initio Quantum Mechanics
- Choose from many commonly-used basis sets (STO-1G to 6-311++g2d2p) including
the standardized STO-3G, 3-21G, 6-31G*, and 6-31G** basis sets
- Extra basis functions ( s, p, d, sp, spd ) can be added to individual atoms or
to groups of atoms.
- Users can define their own basis sets or modify existing basis sets easily
using HyperChem's documented basis set file format.
- Different basis sets can be used on different atoms; use ghost orbitals to
eliminate basis set extension.
- Use electric fields, configuration interaction, MP2, direct SCF and RHF or
UHF.
Density Functional Theory (DFT)
- All the capabilities of HyperChem's
Ab Initio
module, e.g. molecular dynamics, vibrations, etc.
- Any combination of seven exchange potentials (Slater, Hartree-Fock, Becke 88,
Perdew-Wang 91, Gill 96, PBE 96, HCTH 98) and 7 Correlation Potentials (VWN,
Perdew-Zunger 81, Perdew 86, Lee-Yang-Parr, Perdew-Wang 91, PBE 96, HCTH 98).
- Hybrid or Combination Potentials B3-LYP, B3-PW91, EDF1, Becke 97.
Semi-empirical Quantum Mechanics
- HyperChem offers eleven semi-empirical molecular orbital methods, with options
for organic and main-group compounds, for transition metal complexes, and for
spectral simulation.
- Choose from Extended H�ckel,
CNDO, INDO, MINDO/3, MNDO, AM1, RM1, PM3, ZINDO/1, ZINDO/S, and TNDO.
- New Typed Neglect of Differential Overlap (TNDO) method that uses
semi-empirical parameters assigned to atom types rather than atomic numbers
(improved accuracy using ideas from molecular mechanics).
- Applied electric fields (all methods) and magnetic fields (TNDO only).
Molecular Mechanics
- Four force fields provide computationally convenient methods for exploring the
stability and dynamics of molecular systems
- Added flexibility of user-defined atom types and parameters.
- Choose from MM+, a general-purpose force field, and three specialized
biomolecule force fields: Amber, BIO+(Charmm), and OPLS.
- Convenient inspection and editing of all parameters.
Mixed Mode Calculations
HyperChem allows you to perform quantum calculations on part of a molecular
system, such as the solute, while treating the rest of the system classically.
This boundary technique is available for semi-empirical methods and, with some
limits, for ab initio and DFT calculations.
Other Features
Customize and Extend HyperChem with the Chemist's Developer Kit
- Streamline HyperChem's menus. Add new graphical and computational features;
create custom menus for specific applications.
- Interface to Visual Basic, C, C++ and FORTRAN programs. Add dialog boxes as
well as menu items. For example, you could use HyperChem for visualization of
structures and results from non-graphical quantum chemistry programs.
- Link HyperChem procedures to other Windows programs such as MS Word and Excel;
direct selected results to these applications for convenient analysis and
reporting.
- Use HyperChem's custom script editor to interactively execute script commands
or prepare scripts.
- Generate elaborate scripts using the Tcl language and new visual images and
dialog boxes with Tk. These standard languages have been extended with over 700
HyperChem script variables and commands.
Interface HyperChem to a Variety of Third-Party Packages such as GAMESS,
Gaussian, PQS, Q-Chem and Mopac2007
- Open-source interfaces allow anyone to contribute to and/or extend these
interfaces. Interfaces use Visual Studio and C++.
- Interface generates input for third-party package, spawns it and parses its
output to return results to HyperChem.
- Follow the simple model to build a graphical interface to any number-crunching
package of your own.
- All interfaces initially allow a graphical interface to Single Point
calculations for densities and orbitals, Geometry Optimization for display of
structure, and vibrational analysis for display and animation of normal modes.
Manuals, Tutorials, and On-line Help
HyperChem includes a full set of electronic manuals in convenient Adobe Acrobat
format. These manuals are equivalent to six hardcopy manuals (Getting Started,
Reference Manual Vol. 1 and Vol. 2, Modules, Chemist's Developer Kit, and
Computational Chemistry). Separate On-line Help is fully integrated into
HyperChem including Help within the current Context. A rich set of Video
Tutorials is included with HyperChem. Choose from over a hundred tutorials on
various topics in molecular modeling and HyperChem. Sit and listen to our CEO
describe all the feaures of HyperChem.
Licensing Options
A wide variety of licensing options are available with HyperChem including
Hard-Lock (portable dongle - USBt), Soft-Lock (locked to specific machine) and
Network Licensing where a license can be used anywhere on a network. Ask us
about site licensing for your institution or company.
Contact your HyperChem reseller or ask us for a free evaluation disk.
HyperChem is a trademark of Hypercube, Inc. Windows is a trademark of Microsoft
Corporation. All other brand and product names are trademarks of their
respective holders. Printed in the U.S.A.
1115 North West 4th Street Gainesville, Fl 32601 USA Tel: (800) 960-1871 or
(352) 371-7744 Fax: (352) 371-3662 E-mail:
info@hyper.com Web Site:
http://www.hyper.com
Display
- Rendering choices: Ball-and-stick, fused CPK spheres, ball and cylinders, or
tubes with optional shading and highlighting. Also vdW dots added to any
rendering.
- Ribbon rendering for protein backbones, with optional sidechain display.
- Cylinders, ribbon lines, thin solid ribbons, thick ribbons and coils for
secondary structure rendering.
- 3D Isosurfaces or 2D contour plots of: Total charge density. Molecular
orbitals, Spin density, Electrostatic potential (ESP).
- ESP mapped onto 3D charge density surface
- Isosurface rendering choices: wire mesh, Jorgensen-Salem, transparent and
solid surfaces, Gouraud shaded surface. User-specified grid and isosurface
value.
- Generate ray-traced graphical images.
- During simulations, display and average kinetic, potential, and total energy,
as well as values of user-specified bond lengths, bond angles, or torsion
angles.
- Spectra display of IR or UV-VIS.
- Animate vibrational modes.
- NMR spectra.
- Crystal structures.
- Slides (molecules plus annotations).
Customize and Automate
- Construct custom menus
- Automate routine operations with scripts
- Send selected data to files or workspace
- Add new features as menu items, or run from scripts
Interface and Extend
- Construct a custom interface to programs written in VB, C/ C++, or FORTRAN
- Send HyperChem results to MS Word or Excel.
Predict
- Relative stabilities of isomers
- Heats of formation
- Activation energies
- Atomic charges
- HOMO-LUMO energy gap
- Ionization potentials
- Electron affinities
- Dipole moments
- Electronic energy levels
- MP2 electron correlation energy
- CI excited state energy
- Transition state structures and properties
- Non-bonded interaction energy
- UV-VIS absorption spectra
- IR absorption spectra
- Rate constants - unimolecular or bimolecular reactions
- Equilibrium as a function of temperaure
- Isotope effects on vibrations
- Collision effects on structural properties
- Stability of clusters
- Shielding and coupling constants
- Conformations of flexible systems
- Homologous proteins
Save Results
- Use Import/Export option to save results of quantum mechanics calculations or
to view results generated by other programs.
- Use HyperChem Data to store structures and properties in a custom molecular
database.
- Create Reaction Movies in AVI format
Results with HyperChem
System Requirements
A PC running Windows NT, 98, ME, 2000, XP, or Vista. 128 MB of RAM and 50-150 MB
of hard disk space (the requirements for running Windows are generally more
severe than for running HyperChem). Raytracing and using the HTML controls
requires a graphics card with more than 256 colors. Otherwise, any PC graphics
card is acceptable as long as it supports OpenGL.
HyperChem Release 8.0 is available for both standalone and networked
installations. Site licensing is also available; ask us about it!
�@